Síndrome de Ehlers
|
Revisión a proPólito de dos casos con Ehlers subtipo II
|
|
Dra. Médica Zambrano C*.,
Dra. María Eugenia Montesdeoca*, Dra. Alicia Pereira**, Dra
Susana L?ez *** |
|
* Médica R3, Postgrado de Dermatología. Universidad Central
del Ecuador |
|
** Médica Jefe de Servicio de Dermatología Hospital Quito
N?1 |
|
*** Médica Jefe del Servicio de Patología |
|
Hospital Quito N?1 |
|
Hospital Quito N?1 Polic? Nacional |
| |
|
Correspondencia: |
|
Dra. Médica Zambrano C |
|
Pasaje B OE-430 y Pedro Freile, |
|
Quito ? Ecuador. SudaMédica |
Resumen
El síndrome de Ehlers Danlos es una enfermedad poco frecuente
cuyo Diagnóstico es en muchos casos subvalorado. Constituye 11
subtipos con caracteRÃoticas Clínicas propias y comunes. Es de
importancia el realizar un Diagnóstico clínico apropiado descartando
la posibilidad de afectaci? sistópica grave que puede incluso
comprometer la vida del paciente (subtipo IV).
Se hizo una breve descripci? de la patología y se describen dos
casos de transmisión dominante y afectaci? familiar. Por las
caracteRÃoticas Clínicas se los encasill?dentro del subgrupo de
síndrome de Ehlers Dan los tipo II.
Palabras claves: síndrome de Ehlers Danlos subtipo II.
Summary
Ehlers Danlos syndrome is an infrequent disease. Its diagnosis in
many cases is overlooked. There are 11 subtypes and each one has its
own characteristics. It is very important to make a specific
clinical diagnosis, to dismiss the possibility of subtype IV, which
can even endanger the patient?s life. We describe two cases of
dominant transmission and inherited trait with a brief descrIption
of the pathology.
Due to their clinical characteristics they were allocated to the
variant "Ehlers Danlos Type II".
Key words: Ehlers Danlos Syndrome, Ehlers Danlos
Type II
Se adjudica el nombre de síndrome de Ehlers Danlos (SED) a un
grupo heterogóseo de más de 10 asociaciones patolóticas, todas
relacionadas con una alteraci? genótica de la estructura y sÓrtesis
del col?eno y del tejido conectivo(1). El col?eno es una prote?a
altamente distribuida en todo el organismo y su afectaci? se
relaciona con disminución en la cantidad y desorganizaci? de los
haces fibrosos del mismo(5).
En el SED la piel, articulaciones y vasos sanguíaeos son los
óganos principalmente afectados y esto se manifiesta como
hipermovilidad articular, hiperextensibilidad y fragilidad
cutáneas, principalmente.
La incidencia de la enfermedad a nivel mundial es de 1:400000. No
existe preferencia ?nica, aunque el mayor Número de casos
reportados se ha observado en caucúnicos(1).
Al momento se han descrito 11 subtipos de SED y en solo 6 de
ellos se ha identificado el defecto del col?eno el cual está
siempre presente desde el nacimiento aunque las manifestaciones
Clínicas puedan aparecer más tarde. Cada subtipo presenta
diferencias bioqué icas, genóticas y Clínicas
(Tabla 1).
En muchos casos, el SED no se puede clasificar como en los reportes
de anormalidades localizadas en los hombros o en los que existe
anormalidad en la degradaci? intracelular de las cadenas
?
2 del procol?eno tipo I(4), así como en un grupo
de dos familias afectadas con SED y arterias sistópicas
tortuosas(10).
El Diagnóstico es básicamente clínico, tratando de determinar el
subtipo. El Diagnóstico diferencial se debe realizar principalmente
con el pseudoxantoma elÓptico y la cutis laxa (1), aunque también se
debe tomar en cuenta al liquen escleroso atRÃoico, al atrofoderma
perifolicular, y es importante recordar la asociaci? del SED con el
to adelgazamiento díamico(4). síndrome de Marfan y la osteog?esis
imperfecta(3). En muchos casos, la biopsia y el estudio
histopatolúnico pueden reportar normalidad. Los resultados publicados
son contradictorios. Los estudios recientes establecen que la dermis
es normal vista por el microscopio Óptico, excepto en las variantes
IV y VI en las que se ha descrito.
No existe un tratamiento satisfactorio. Un estudio revel?que en
pacientes con SED tipo IV hubo respuesta favorable al uso de
vitamina C a dosis elevadas (4 g/día) con disminución del sangrado y
mejor cicatrizaci? de heridas(1).
Caso clínico 1
Mujer de 37 años de edad, casada, procedente de la provincia del
Cotopaxi, con antecedentes familiares de padre y abuelo paterno
portadores de enfermedad cutárea similar la que cursa la paciente.
Nacida por parto normal de un embarazo a término. Desde la infancia
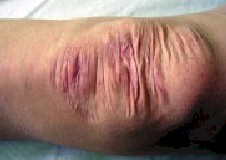
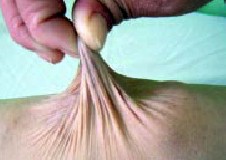
presenta fragilidad e hiperextensibilidad cutáneas (Foto 3),
aumento de la flexibilidad articular sobre todo a nivel de
articulaciones interfalérgicas (Foto 2). Al examen físico se puede
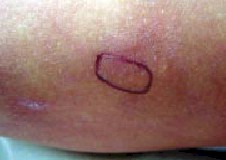
observar pseudotumoraciones de aproximadamente 0,5 a 1 cm de di?etro
(Foto 4), movibles, con superficie cutárea normal. más tiples
cicatrices atRÃoicas con la caracteRÃotica en "papel de
cigarrillo" (Foto 1), localizadas sobre todo en miembros inferiores
y Áreas de roce.
 |
 |
|
Fig.1 Cicatrices atRÃoicas en papel de
cigarrillo |
Fig. 2 Hiperflexibilidad articular |
| |
|
 |
 |
|
Fig. 3. Hiperextensibilidad cutárea |
Fig 4. Pseudotumores de peque? tamaño |
Caso clínico 2
Ni? de 7 años de edad, hija de la paciente del caso previamente
descrito. Nacida por parto normal, a las 28 semanas de gestación. Al
igual que la madre, desde el nacimiento presenta caracteRÃoticas
Clínicas similares. En la ni? se observa además epicanto
importante y lenta cicatrizaci? de heridas. El desarrollo neurológico
en ambos casos ha sido totalmente normal.
El examen cardiolúnico (electrocardiograma y eco car-díaco) a ambas
pacientes(1) report?normalidad. El chequeo oftalmolúnico
detect?Hipermetropía y Miopía en la ni? para lo cual usa lentes
correctivos.
La biopsia de piel report?alteraci? leve a nivel de fibras
elópticas. Se solicit?estudio genúnico para determinar el subtipo
especúnico de este síndrome; sin embaraso, el examen no se hizo por
no haber disponibilidad econótica para el mismo.
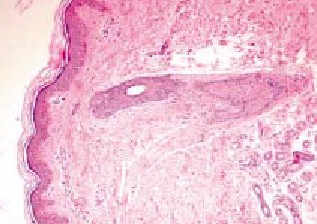
Fig. 5 Histopatología sin evidencia de mayores cambios en fibras
elópticas
Discusi?
El síndrome de Ehlers Danlos es una patología poco frecuente y
con la ventaja de que en la mayoRÃo de los diferentes subtipos el
pronÓptico es favorable. Adjudicamos a este hecho el desconocimiento
de la verdadera incidencia de la enfermedad en nuestro medio, pues
al ser una entidad que en la mayoRÃo de casos no afecta la calidad
ni forma de vida del paciente, ?te no recurre al médico para
determinar el Diagnóstico o la probabilidad de un tratamiento.
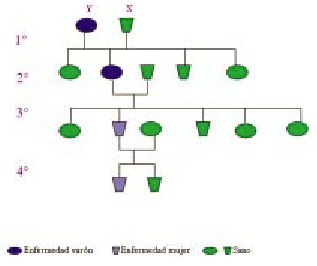
La mayoRÃo de las variantes se transmiten en forma dominante
pero en pocos casos es evidente la manifestaci? Clínica familiar.
Por ello consideramos importante la presentaci? de dos casos: madre
e hija (tercera y cuarta generaciones familiares) con el reporte
verbal de manifestaciones Clínicas similares en dos lÁreas
familiaresprevias.
Todos los signos clínicos observados en ambas pacientes encuadran
dentro del síndrome de Ehlers Danlos tipo II, sin dejar la
probabilidad abierta de que podrÃa n pertenecer a la subclase I,
aunque consideramos que la hiperelasticidad cutárea e
hiperextensibilidad articular son más graves en esta variante (I).
Ni siquiera el exa-men genúnico habRÃo logrado despejar esta duda,
pues las alteraciones genóticas son similares en ambos casos. La
verdadera importancia de este estudio estaRÃo más bien orientada a
conocer la probabilidad de que en un siguiente embarazo el producto
curse o no con la enfermedad.
Lastimosamente no podemos brindar posibilidad de tratamiento,
solo indicar a las pacientes la importancia del cuidado de la piel
al evitar cualquier tipo de traumatismo y dejar claro que por las
caracteRÃoticas de transmisión familiar observadas, es alta la
probabilidad de que una nueva descendencia manifieste la enfermedad.
?
|
 |
|
Fig. 5 Histopatología sin
evidencia de mayores cambios en fibras elópticas |
| |
|
 |
|
Cuadro de transmisión familiar |
Referencias
1. Ceccolini E, Schwartz R. Ehlers-Danlos Syndrome. 2002, April
9. URL: https://www.emedicine.com
2. Fitzpatrick T, Eisen A, Wolff K y col. Dermatología en Medicina
General; Trastornos hereditarios del tejido conectivo con cambios
cutóseos: Síndrome de Ehlers Danlos. Editorial Panamericana. 4ta
edici?. Buenos Aires Argentina, 1997:p. 2050-52
3. Iglesias D. Tratado de Dermatología. Síndrome de Ehlers Danlos;
Madrid, Espaís, 1994:416
4. Weedon D. Piel Patología: Síndrome de Ehlers Danlos. Ed. Marban;
Madrid. Espaís, 2002:p. 304-306
5. Ratajczak C, Bielenzka L. Collagens, the basic proteins of the
human body; J Appl Genet. 2000;41(4):317-30
6. Mao J, Bristow J. The Ehlers-Danlos Syndrome: Beyond
collagens; J Clin Invest: 2001 May;107(9):1063-69
7. Nuytinck L, Freund M, Lagae L, et al. Classical Ehlers-Danlos
syndrome caused by a mutation in type I collagen; Am. J. Hum. Genet.
2000;66:1398-402
8. Hussain A, Zeisberger S., Hubber P, et al. Brittle cornea
syndrome and its delineation from the kyphoscoliotic type of
Ehlers-Danlos syndrome (EDS VI): report on 23 patients and review of
the literature. Am J Med Genet. 2004 Jan 1;124A(1):28-34
9. Fichard A, Chanut-Delalande H, Ruggiero F. [The Ehlers-Danlos
syndrome: the extracellular matrix scaffold in question]. Med Sci
(Paris). 2003 Apr;19(4):443-52
10. Abdul A, Janahi IA, Eltohami A, et al. A new type of
Ehlers-Danlos syndrome asso-ciated with tortuous systemic arteries
in a large kindred from Qatar. Acta Paediatr.
|
|
|
|
| |
|
|
|
|
|